

In yet another example of how computation is transforming biology and medicine, Allen School researchers have developed a machine learning-based system that improves upon a widely used technique for analyzing interactions between DNA and the proteins that regulate gene expression. In a paper published in Nucleic Acids Research, Ph.D. students Nao Hiranuma and Scott Lundberg and professor Su-In Lee demonstrate how their system, AIControl, is more cost-effective — and yields more accurate results — than current practices for increasing our understanding of genetic factors regulating the onset of disease and other biological processes.
AIControl is designed to be used in conjunction with chromatin immunoprecipitation and DNA sequencing (ChIP-seq), a vital tool in molecular biology for determining the location and function of transcription factors that govern gene expression. ChIP-seq enables researchers to map the binding sites of a specific regulatory protein to DNA across the human genome. While ChIP-seq is one of the most advanced and popular techniques available, it is not without its shortcomings; in addition to being a costly experiment to run, the data generated by immunoprecipitation (IP) contains background signals that can lead to false positives.
To compensate, researchers are advised to generate an additional control dataset in addition to their target dataset. While the IP target dataset captures actual protein binding signals, the control captures potential biases in the data. The results of both are then subjected to a process known as “peak calling,” in which algorithms compare the two datasets and separate out the peaks, which indicate the presence of true protein binding signals, and minimize false positives stemming from background noise. It is these peaks that researchers are interested in exploring, as they indicate the site of DNA-protein interactions that influence biological processes.
Due to the time and expense associated with generating that second dataset, many users opt to rely on an existing control pulled from a public database or forego the recommended control altogether. As an alternative, the Allen School team developed a machine-learning framework, AIControl, that replaces the need for the additional control dataset by estimating it in silico using multiple, publicly available controls.

AIControl works by systematically determining the most appropriate combination of control datasets to be applied to the target experiment, then estimates the distribution of unwanted background signals based on those datasets to identify the true binding peaks. With AIControl, researchers are able to rely on efficient and cost-effective computation, rather than expensive biological experiments, to generate more accurate ChIP-seq results.
“By making use of existing datasets on a large scale, AIControl can save researchers time and expense while offering a more comprehensive and accurate peak analysis of their target dataset,” explained Hiranuma. “Because our system is capable of leveraging information from a large number of control experiments in a public database, AIControl captures potential biases in the data that might be missed using a single control — or using no control at all.”
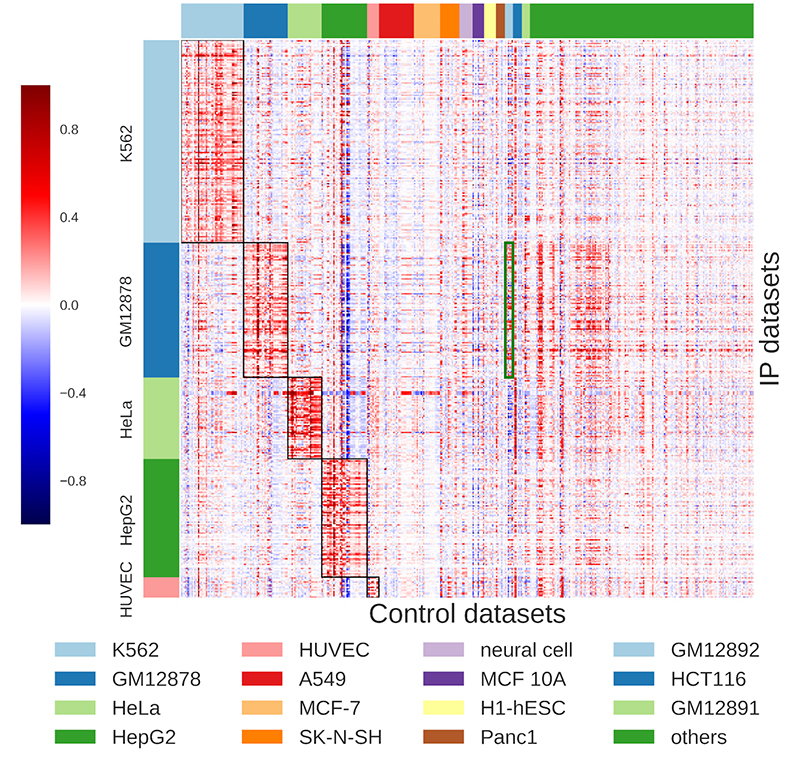
While existing peak calling techniques require the user to decide which control datasets to apply, AIControl alleviates that burden by automatically integrating and weighing multiple datasets that are most relevant to the target dataset. The system draws upon data from 440 publicly available controls, encompassing more than 100 cell types, to infer the distribution of background signals for the peak calling comparison. Hiranuma and his colleagues evaluated their system by applying AIControl to 410 IP datasets from the ENCODE ChIP-seq database spanning five major cell types.

AIControl outperformed existing peak calling methods in identifying putative binding sites, including in cases where control datasets of the same cell type were removed. This suggests that AIControl will be capable of reliably estimating background signals in conjunction with ChIP-seq analyses performed on new cell types. According to Lee, the team’s findings have already generated interest among biotechnology companies eager to replace a costly process for generating new data with an AI-driven solution.
“Locating the binding sites of regulatory proteins on DNA is a central problem in molecular biology that will enable us to more fully understand the interplay between genetic factors and disease,” Lee said. “With AIControl, we have shown that machine learning can be used in place of expensive biological experiments to generate results with greater speed and accuracy than standard approaches. Our hope is that this will advance our understanding of genetic factors that influence disease and, ultimately, lead to better outcomes for people.”
Read the journal paper here, and visit the project webpage here.